Vigilancia epidemiológica basada en secuenciación de genoma completo de SARS-CoV-2 en Ciudad de México y área metropolitana
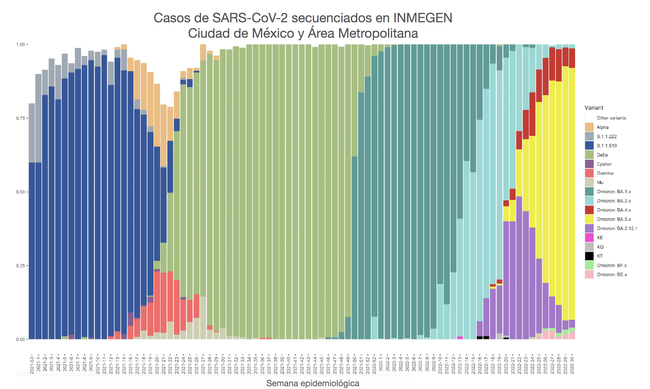
Figura 1. Figura 1. Prevalencia de variantes de SARS-CoV-2 secuenciadas por INMEGEN en Ciudad de México y área metropolitana a través del tiempo.
Generalidades de SARS-CoV-2
A finales de 2019, una serie de casos de neumonía de causa desconocida fueron ligados epidemiológicamente a un mercado en Wuhan, Hubei, China [1] El subsecuente análisis, utilizando tecnología de secuenciación de última generación, reveló la existencia de un nuevo coronavirus identificado como coronavirus de tipo 2 causante del síndrome respiratorio agudo severo (SARS-CoV-2) por el Comité Internacional de Taxonomía de Virus (ICTV); la enfermedad causada por este nuevo virus se denominó COVID-19 [2]. Hasta agosto de 2021, el virus se ha dispersado por todo el mundo, con 200 millones casos confirmados y 4.2 millones de decesos, en México se han reportado 2.89 millones de casos confirmados y 242 mil muertes [3].
Vigilancia epidemiológica basada en secuenciación de SARS-CoV-2
Las mutaciones en el genoma de SARS-CoV-2 pueden resultar en cambios en el comportamiento biológico y clínico del virus, estos cambios afectan su transmisibilidad, patogencidad y su respuesta ante el sistema inmunológico, pudiendo disminuir la eficacia en de la neutralización por anticuerpos terapéuticos, por vacunas o generados de forma natural después de una infección [4, 5]. En el caso del SARS-CoV-2, las proteínas virales acumulan mutaciones con tasas diferentes, en particular, las mutaciones en la proteína espiga tienen un papel biológico importante, dada la relevancia de esta proteína en el mecanismo de ingreso a la célula hospedera durante una infección [6, 7].
La mejor forma de identificar, monitorear y determinar el grado de dispersión de las variantes virales es mediante el establecimiento de programas de vigilancia epidemiológica de base genómica, en las cuales se apliquen tecnologías de secuenciación de ácidos nucleicos que permitan analizar la secuencia completa del genoma viral con la finalidad de detectar los perfiles mutacionales que caracterizan a cada variante.
En este sentido, la OMS publicó el 8 de enero del 2021 el documento: “Genomic sequencing of SARS-CoV-2: a guide to implementation for maximum impact on public health”, donde exhorta a los países miembros de la organización a establecer programas de vigilancia epidemiológica basadas en secuenciación y hace una serie de recomendaciones técnicas y operativas para que estos programas resulten en un impacto real en la salud pública [8].
Los esfuerzos internacionales por la secuenciación de muestras virales en pacientes COVID-19 comenzaron en diciembre de 2019. Las principales bases de datos que recopilan esta información son GISAID (Global Initiative on Sharing Avian Influenza Data), NCBI (National Center for Biotechnology Information) y ViPR (Virus Pathogen Resource).
Secuenciación de SARS-CoV-2 en la CDMX
El INMEGEN estableció un programa de vigilancia genómica del virus SARS-CoV-2 desde agosto del 2020 en el Valle de México y desde mayo de 2021 en el sureste del país. Se establecieron protocolos de secuenciación de genoma completo de SARS-CoV-2 con dos plataformas: secuenciación con nanoporos (Oxford Nanopore) y secuenciación por síntesis de Illumina.
La primera secuencia de genoma completo con la que el INMEGEN contribuyó a la plataforma pública de datos GISAID fue depositada el día 26 de agosto del 2020 y actualmente el INMEGEN ha secuenciado y depositado en en 23,045 genomas de SARS-CoV-2, de los cuales 15,603 pertenecen a la Ciudad de México y Estado de México y 6,219 al interior del país. Estos casos secuenciados provienen de pacientes atendidos en las Jurisdicciones Sanitarias de la CDMX, de la Unidad Temporal COVID-19 de CITIBANAMEX, del Hospital Ajusco Medio, del Instituto Nacional de Ciencias Médicas y Nutrición "Salvador Zubirán" y de laboratorios de la red de Salud Digna.
Panorama actual de la presencia de variantes
Hasta finales del mes de marzo del 2021 la variante B.1.1.519 representaba el 86% de los genomas secuenciados. A partir de la segunda semana de abril, esta variante comienza a disminuir llegando al 5% en la segunda semana de Junio. Las variantes de preocupación (Alfa, Gamma y Delta) comienzan a aumentar a partir de la tercera semana de abril,y en la tercera semana de noviembre del 2021 la variante Ómicron BA.2 representó el 87.5% y Ómicron BA.1 el 12.5%. Para más detalle, consultar la figura 1.
Se presenta la distribución de las variantes de Ómicron que circulan en las semanas epidemiológicas 28 y 29.
| CDMX y Área Metropolitana | |||
|---|---|---|---|
| Semana | Variante | Casos secuenciados | Porcentaje |
| 2022-29 | Omicron: BA.1.x |
0 |
0% |
| Omicron: BA.2.x | 6 | 1% | |
| Omicron: BA.4.x | 36 | 6% | |
| Omicron: BA.5.x | 492 | 86% | |
| Omicron: BA.2.12.1 | 18 | 3% | |
| Omicron: BF.x | 9 | 2% | |
| Omicron: BE.x | 10 | 2% | |
| 2022-30 | Omicron: BA.1.x | 0 | 0% |
| Omicron: BA.2.x | 1 | 1% | |
| Omicron: BA.4.x | 5 | 7% | |
| Omicron: BA.5.x | 64 | 85% | |
| Omicron: BA.2.12.1 | 2 | 3% | |
| Omicron: BF.x | 1 | 1% | |
| Omicron: BE.x | 2 | 3% | |
*Quintana Roo, Yucatán, Tabasco, Guerrero y Oaxaca
- Zhu, N., et al., A Novel Coronavirus from Patients with Pneumonia in China, 2019. N Engl J Med, 2020. 382(8): p. 727-733.
- Coronaviridae Study Group of the International Committee on Taxonomy of, V., The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol, 2020. 5(4): p. 536-544.
- WHO, WHO Coronavirus (COVID19) Dashboard. 2021. https://www.worldometers.info/coronavirus/
- Korber, B., et al., Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell, 2020. 182(4): p. 812-827 e19.
- Plante, J.A., et al., Spike mutation D614G alters SARS-CoV-2 fitness. Nature, 2020.
- Guruprasad, L., Human SARS CoV-2 spike protein mutations. Proteins, 2021.
- Li, Q., et al., The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity. Cell, 2020. 182(5): p. 1284-1294 e9.
- WHO, Genomic sequencing of SARS-CoV-2: a guide to implementation for maximum impact on public health. 2021.
|
|
|
|
||
La legalidad, veracidad y la calidad de la información es estricta responsabilidad de la dependencia, entidad o empresa productiva del Estado que la proporcionó en virtud de sus atribuciones y/o facultades normativas.